
5 Pituitary Neoplasia and Pathology
Ronald M. Lechan, MD, PhD and Arthur Tischler, MD
Learning Objectives
- To classify pituitary adenomas (now classifed as PitNets) and appreciate that non-secreting adenomas and prolactinomas are the most common pituitary adenomas
- To describe the mechanisms underlying the development of pituitary adenomas
- To form a differential diagnosis of hyperprolactinemia and appreciate that not all hyperprolactinemia is due to a prolactinoma
- To identify the clinical manifestations of hyperprolactinemia including prolactinoma
- To recall pathogenetic mechanisms and clinical manifestations of somatotroph adenoma (Acromegaly)
- To discuss the steps in the diagnostic algorithm of hypercortisolism.
- To describe the clinical manifestations of corticotroph adenoma (Cushing’s disease)
- To discuss the pathogenesis and clinical manifestations of non-secreting pituitary adenoma
- To decide on an appropriate treatment course with surgery and medical therapies for pituitary neoplasms based on different clinical characteristics
- Describe the differential diagnosis of functional and non-functional pituitary masses and recognize the major pathological findings.
Key Concepts
- The vast majority of pituitary neoplasms are benign adenomas with non-secreting adenomas being most common. Treatment may not be needed unless causing local symptoms such as compression of the optic chiasm or cavernous sinus invasion with cranial nerve deficits.
- In addition to pituitary tumors, hyperprolactinemia may be related to pregnancy, several medications especially those which block dopamine, primary hypothyroidism, liver disease, kidney disease and other situations.
- Prolactinomas are the most common secretory adenomas and are usually treated medically with dopamine agonists.
- Acromegaly is uncommon but can cause an increase in mortality. IGF-1 level may be diagnostic if elevated for age and sex. A glucose tolerance test to assess for GH suppression is often done for confirmation. Treatments include surgery, radiation, somatostatin analogues and a GH antagonist.
- Cushing’s syndrome can be subtle in most cases but can lead to significant morbidity due to the chronic effects of cortisol excess on the body.
- If one has a clinical suspicion for Cushing’s syndrome based on history and physical, initial testing may include a 1 mg dexamethasone suppression test, 24 hour urinary cortisol or nighttime salivary cortisol. If initial testing suggests Cushing’s, an ACTH level should be done. To differentiate Cushing’s disease from a pituitary adenoma from ectopic ACTH production, testing may include pituitary imaging, high dose dexamethasone testing, CRH testing and inferior petrosal sinus sampling.
- Treatment of Cushing’s disease may include surgery, radiation and medications to inhibit cortisol synthesis or ACTH secretion.
- Pituitary adenomas arise in the adenohypophysis (or rarely in pharyngeal pituitary remnant), are homogeneous and usually grow in diffuse sheets.
- H&E staining of pituitary adenomas can be acidophil, basophil or chromophobe, function mirrors that of the normal cell types
- Crooke’s hyaline change is a circumferential perinuclear cytoplasmic clearing caused by accumulation of cytoplasmic filaments in ACTH-producing basophils and indicates feedback inhibition by high levels of glucocorticoids.
- Craniopharyngiomas are non-secretory, often suprasellar and consist of architecturally complex arrangements of squamous, cuboidal, and columnar cells and keratinous debris.
- Architectural complexity usually distinguishes tumor from cyst.
Introduction
Pituitary hypersecretion may occur for several reasons. It may occur physiologically, for example when LH and FSH rise during menopause due to loss of feedback inhibition by gonadal steroids, or when ACTH rises during stress; as a result of hyperplasia of one or more cell types in the anterior pituitary due to the hypersecretion of hypothalamic releasing factors from tumors in the hypothalamus (hamartomas, gangliocytomas) or in the periphery (carcinoids, islet cell tumors), or arise directly from pituitary adenomas. The following will focus exclusively on pituitary adenomas, the most common cause for pathologic hypersecretion of the anterior pituitary.
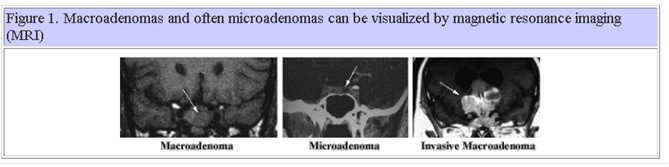
Pituitary adenomas arise from anterior pituitary cells and therefore, are almost always located in the sella turcica, the bony recess of the sphenoid bone in the middle cranial fossa. They comprise approximately 10% of all intracranial neoplasms. Adenomas that are less than 1 cm in diameter are commonly referred to as microadenomas, whereas adenomas that are 1 cm or more are commonly referred to as macroadenomas. Macroadenomas and often microadenomas can be visualized by magnetic resonance imaging (MRI) as shown in Figure 1. Radiologic imaging techniques cannot identify the type of pituitary adenoma. A subgroup of macroadenomas is classified as invasive due to their massive size and invasion of local structures. Pituitary adenomas are common disorders as up to 27% of individuals at autopsy are found to have one. It can be assumed, therefore, that the majority of pituitary adenomas go unrecognized during life and only those that result in endocrine syndromes as a result of their hypersecretion or cause problems as a result of local growth and compression of adjacent structures or hypopituitarism, come to medical attention.
 Pathogenesis of Pituitary Neoplasia
Pathogenesis of Pituitary Neoplasia
Virtually all pituitary adenomas are clonal, i.e. they arise from a single cell. This indicates that the cause for adenoma formation arises from a genetic alteration intrinsic to the anterior pituitary. This alteration may occur as a result of single point mutations that result in excess gene expression (activating mutations) or disruption of genes that are involved in the suppression of cell proliferation (inactivating mutations). For example, in approximately 30 – 40% of Caucasians with somatotroph adenomas (acromegaly), the pathogenesis is believed to be secondary to an activating mutation involving the stimulatory G proteins (Gs). Missense mutations replacing residue 201 (Arg to Cys or His) or 227 (Gln to Arg or Leu) result in ligand-independent constitutive activation of the GHRH receptor and thereby cAMP, which may contribute to the growth and hypersecretory state of these adenomas. Inactivating mutations may be responsible for the increased incidence of pituitary adenomas in the multiple endocrine neoplasia type I syndrome (MEN-I), an autosomal dominant genetic disorder due to a mutation of the menin gene on chromosome 11 that is believed to be a tumor suppresser gene. Excess secretion of basic fibroblast growth factor (FGF-2) and a novel pituitary tumor transforming gene product, PTTG, are also thought to have an early role in pituitary cell transformation. The majority of pituitary adenomas are benign. Rarely, malignant transformation of a pituitary adenoma occurs, which may be associated with metastasis both within and outside of the CNS, likely secondary to a second mutation that may include ras and p53 mutations as late events.
Etiology of Pituitary Adenomas
Pituitary adenomas (PitNETs are probably best classified by their hypersecretory product. It must be understood, however, that some tumors secrete more than one pituitary hormone (plurihormonal adenomas). Often pituitary adenomas may synthesize one or more of the classic anterior pituitary hormones. Some pituitary adenomas are incapable of secretion or may secrete the alpha- or beta subunits of the glycoprotein hormones (LH, FSH, TSH) that might otherwise go unnoticed due to their biologic inactivity. Table 1 lists the most common types of pituitary adenomas, their most abundant secretory products and their relative frequency.
| Table 1. Classification of Pituitary Adenomas | ||
| Tumor Type | Secretory Product(s) | Relative Frequency (%) |
| Prolactinoma (Lactotroph Adenoma) | Prolactin | 50 |
| Somatotroph Adenoma | Growth Hormone/Prolactin | 10 |
| Corticotroph Adenoma | ACTH | 5 |
| Thyrotroph Adenoma | TSH | 1 |
| Nonsecreting Adenoma | α-subunit | 34 |
Lactotroph Adenoma (Prolactinoma)
The prolactinoma is the most common pituitary adenoma. In females, the majority of prolactinomas present as microadenomas whereas in males, the majority present as macroadenomas. This does not necessarily indicate that microadenomas become macroadenomas. In fact, several studies have demonstrated that in females, microadenomas remain microadenomas, even when followed for as long as 15 years. Thus, prolactinomas comprise part of a group that may have different biologic behaviors. Symptoms associated with hyperprolactinemia are listed in Table 2. Amenorrhea and impotence are due to the effect of prolactin to inhibit GnRH secretion with the subsequent reduction in LH/FSH secretion, as well as interference with the actions of LH/FSH at the gonads. The result is a fall in estrogen or testosterone levels. Prolactin may also stimulate androgen secretion from the adrenal gland, resulting in hirsutism. Because microadenomas are more commonly found in females, symptoms due to compression of local structures are generally found in males.
| Table 2. Clinical Manifestations of Hyperprolactinemia | |
| Females | Males |
| Galactorrhea (found in about 30 – 80% of women) |
Galactorrhea (found in less than < 10% of men) |
| Amenorrhea | Impotence |
| Infertility | Hypogonadism |
| Hirsutism | Visual Field Abnormalities |
| Extraocular Muscle Palsies | |
| Headaches | |
Unless the plasma prolactin level is greater than 200 ng/ml (Normal < 20 ng/ml), other etiologies should be considered as the cause for the hyperprolactinemia. These are listed in Table 3.
| Table 3. Etiologies of Hyperprolactinemia (adapted from M.E. Molitch, Endocrinology and Metabolism Clinics of North America, 28, 143-169, 1999) | ||
| Pituitary Disease | Hypothalamic Disease | Medication |
| Prolactinoma Acromegaly Nonsecreting Adenoma Empty Sella Syndrome Lymphocytic Hypophysitis |
Craniopharyngioma Meningioma Dysgerminoma Histiocytosis Sarcoidosis Stalk Section |
Phenothiazines Haloperidol Reserpine MAO Inhibitors Tricyclic Antidepressants Methyldopa Metoclopramide Cocaine Verapamil Fluoxetine |
| Neurogenic | Other | |
| Herpes Zoster Chest Wall Lesions Spinal Cord Lesions Breast Stimulation |
Pregnancy Hypothyroidism Renal Failure |
|
In the normal pituitary gland, prolactin release is primarily under inhibitory regulation by dopamine (Figure 2). Thus, anything that disconnects the hypothalamus from the anterior pituitary or disrupts blood flow from the hypothalamus to the anterior pituitary will cause hyperprolactinemia. Decreased metabolism of prolactin, as may occur in renal failure, or medications that antagonize the dopamine D2 receptor commonly used for psychiatric disorders, will also cause hyperprolactinemia. Finally, a number of peptides intrinsic to the hypothalamus including TRH (thyrotropin-releasing hormone) and VIP (vasoactive intestinal polypeptide) act as prolactin-releasing factors. Stimuli that increase these peptides such as hypothyroidism or breast stimulation, respectively, will also cause the release of prolactin. Since the majority of women with prolactinomas have prolactin levels below 150 ng/ml, distinguishing a prolactinoma from other causes for hyperprolactinemia is particularly important.
| Figure 2. Physiology of Prolactin Secretion | |
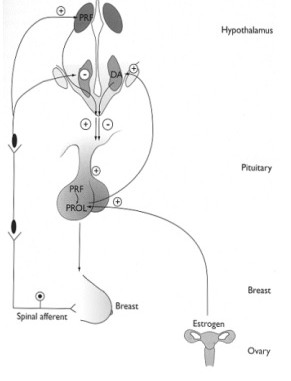 |
Prolactin is primarily under tonic inhibitory control by the hypothalamus through the release of dopamine (DA). Several prolactin-releasing factors (PRFs) have been identified in the hypothalamus that may play a role under certain conditions such as suckling or stress. In the case of suckling (or breast stimulation), tonic inhibition of prolactin secretion is overridden by neurogenetic signals from the breast through a multisynaptic pathway that stimulates PRF and inhibits dopamine. Estrogen also induces hyperplasia and hypertrophy of prolactin cells and increases prolactin secretion. |
Treatment of patients with prolactinomas depends upon the size of the adenoma and symptoms. If the adenoma is small and symptoms are minimal, observation may be all that is necessary since the majority of these adenomas will not enlarge. Some adenomas that require therapy respond medically to the dopamine agonists, bromocriptine or cabergoline. By stimulating dopamine D2 receptors on the adenoma, these drugs cause a reduction in tumor size and inhibit prolactin synthesis and secretion. Cabergoline may also have additional apoptotic effects. Morphologically, treated tumors show involution of rough endoplasmic reticulum, decreased cytoplasmic volume, and reduced secretory vesicles. If resistant to medical therapy, transsphenoidal surgery is performed. Surgical success rates are high for microadenomas (>70%) but postoperative recurrence may be as high as 50% over 5 years. Macroadenomas are more difficult to cure by surgery with a success rate of only ∼30%. Radiotherapy is not very effective and is only used as a last resort.
Somatotroph (GH) Adenoma
Hypersecretion of growth hormone (GH) results in gigantism in children and adolescents before closure of the epiphysis, and acromegaly in adults. GH is a 191 amino acid protein secreted in a pulsatile fashion, every 2-4 hours. There is also a diurnal variation to GH secretion such that more GH is secreted during the night than during the day. GH secretion is under hypothalamic control, as shown in Figure 3. GH secretion is under the regulation of two hypothalamic peptides, GH releasing hormone (GHRH), which stimulates GH secretion, and somatostatin, which inhibits GH secretion.
| Figure3. Physiology of GH Secretion | |
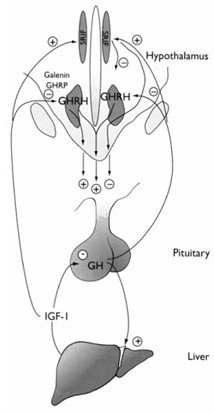
|
GH is primarily under the regulation of two opposing regulatory peptides, GHRH and somatostatin. GHRH stimulates GH transcription and secretion by activating c-AMP, whereas somatostatin inhibits GH secretion by reducing cAMP. GH stimulates IGF-1, and IGF-1 then exerts feedback effects on the pituitary to inhibit GHRH secretion. GH also exerts short feedback effects directly on somatostatin neurons to increase its secretion and on GHRH neurons to inhibit GHRH. Reciprocal inhibitory connections between GHRH and somatostatin neurons may further contribute to the regulation of GH secretion |
It is important to recognize that the actions of GH are only partly due to direct effects of this protein. Many of the actions of GH are mediated by insulin-like growth factor-1 (IGF-1), formally called somatomedin C, which is secreted primarily by the liver (but also other tissues) under the influence of GH. The actions of GH and IGF-1 are listed in Table 4. Because of the pulsatile nature of GH and its short half-life in the circulation (20-25 minutes) compared to the constant levels of IGF-1, measurement of random GH levels are generally not helpful in the diagnosis of acromegaly. Indeed, nighttime GH levels at the peak of a GH spike can be well within the range of an individual with acromegaly. Conversely, normal GH levels may also be found in individuals with acromegaly but these individuals lose the normal pulsatile rhythm of GH.
| Table 4. Physiological Effects of GH and IGF-1 | |
| GH | IGF-1 |
| Insulin Resistance | Enhanced Protein Synthesis |
| Lipolysis | Stimulation of Skeletal Growth |
| Stimulate IGF-1 | Cell Proliferation |
| Immunomodulation | |
GH hypersecretion is almost always due to a pituitary adenoma. Extra-pituitary GH excess is exceedingly rare. Excess GH secretion may also result from hyperplasia of anterior pituitary somatotrophs as a result of ectopic secretion of GHRH (from carcinoid tumors or other neuroendocrine tumors, small cell lung cancer) or due to the genetic disorder, McCune-Albright syndrome (characterized by polyostotic fibrous dysplasia, pigmented skin patches, endocrine abnormalities). At least two genetic mutations are known to give rise to GH-secreting pituitary adenomas, an activating mutation involving the alpha subunit of the stimulatory G gene (∼40%), resulting in ligand-independent constitutive activation of the GHRH receptor, and a germline mutation in the aryl hydrocarbon receptor-interacting (AIP) protein.
Acromegaly has a peak incidence between 40 and 50 years. Similar to other pituitary adenomas, the manifestations of acromegaly are due to the functional consequences of GH hypersecretion and direct mass effects. Acromegaly leads to profound physical deformity due to skeletal overgrowth (bone and cartilage) and fibroblast proliferation under the influence of IGF-1. This is particularly manifest in the facial bones, where there may be enlargement of the mandible, resulting in protrusion of the jaw (prognathism), dental malocclusion, and increased spacing between the teeth. The facial features are thickened, coarsened, or swollen, making skin creases very pronounced, partly due to sodium and water retention and increased glycosaminoglycan accumulation in the skin. The lips and tongue become very large. Increased cartilaginous growth contributes to the very prominent nose. Enlargement of the hands and feet result in the common complaint that ring, glove and shoe size increase. Arthralgias are particularly common (75%) due to cartilaginous overgrowth in the joints resulting in misalignment and destabilization of the joints and ultimately joint destruction. Other manifestations of acromegaly are listed in Table 5. If untreated, acromegaly is associated with a 2 to 3-fold increased mortality due to cardiovascular disease (hypertension, cardiomyopathy, arrhythmias, stroke), cancer (adenocarcinoma of the colon), and respiratory impairment. It is important to make the diagnosis of acromegaly early to prevent many of the devastating complications including premature mortality. Many of the manifestations of soft tissue overgrowth can be reversed, at least in part, by treatment.
Table 5. Manifestation of Acromegaly (adapted from A.G. Harris, Acromegaly and Its Management, Lippincott-Raven, 1996)
| General | Cardiovascular | Gastrointestinal |
| Soft Tissue Swelling | Hypertension | Colonic Polyps |
| Acral Enlargement | Cardiac Enlargement | Enlarged Colon |
| Musculoskeletal | Respiratory | Neurologic |
| Arthralgia | Tongue Enlargement | Paresthesias |
| Prognathism | Sleep Apnea | Carpel Tunnel Syndrome |
| Somnolence |
| Cutaneous | Pyschological | Metabolic |
| Increased Sweating | Depression | Hyperglycemia |
| Acne | Decreased Vitality | Hyperlipidemia |
| Skin Tags | Hypercalcuria |
Elevated IGF-1 concentration suggests the diagnosis of GH excess but IGF-1 can be elevated in physiologic conditions such as puberty or pregnancy and falsely low in liver disease, renal disease, malnutrition or with exogenous estrogen administration. The diagnosis of acromegaly is confirmed by demonstrating that GH secretion cannot be suppressed below 1 ng/ml with a glucose load (oral glucose tolerance test, OGTT). One could also establish the diagnosis by frequent sampling of GH and demonstrating the absence of normal pulsatile GH secretion, but this is time consuming and costly and not readily available in clinical practice.
The majority (over 70%) of patients with acromegaly have macroadenomas (>1 cm), due to the insidious nature of the disease and delay in diagnosis. The goal of therapy is to control GH hypersecretion and reduce tumor volume. Trans-sphenoidal surgery to remove the tumor is the treatment of choice for most patients. Following surgery, GH assessment should be done with measurement of IGF-1 as well as repeat OGTT. Microadenomas can be removed by transsphenoidal surgery with an approximately 80% success rate. Less than 50% of individuals with macroadenomas achieve a surgical cure. Preoperative GH levels are inversely related to surgical success. Several medical treatments (shown in table 6) have recently become available to control the GH hypersecretion and/or the effects of GH in the periphery. These medications can be used as primary therapy in individuals who have macroadenomas to control GH hypersecretion, but generally used as secondary therapy following trans-sphenoidal surgery, if GH hypersecretion persists. Radiation therapy is only partly successful in reducing GH hypersecretion to normal, although preliminary data using gamma knife therapy has suggested an approximately 60% cure over 4 years.
| Table 6. Medical Therapy for Acromegaly | |||||||
| Category | Drug (Trade Name) |
Mechanism of Action | Half life | Administration | Main Adverse Effects | Other Features of Concerns | Cost, per month |
| Ergot alkaloid | Bromocriptine (Parlodel) |
Bind to the dopamine D2 receptor. Mechanism unknown, presumably a paradoxical inhibition of dopamine on GH secretion from the adenoma. | 5-10 hours | Orally daily | Nausea/Vomiting Orthostatic hypotension |
Headache Depression Psychosis Fatigue Digital vasospasm |
$62 |
| Cabergoline (Dostinex) |
70 hours | Orally twice weekly |
Nausea/Vomiting Orthostatic hypotension Cardiac valve abnormalities (rare) |
Headache Depression Psychosis Fatigue Digital vasospasm |
$252 | ||
| Somatostatin analogs | Octreotide (Sandostatin) |
Inhibits GH secretion by binding to somatostatin 2 and 5 receptors, providing therapeutic specificity | 2-3 hours | SC injection three times daily |
Diarrhea Biliary sludge/stones |
Hyperglycemia | $1,500 |
| Octreotide-LAR (Sandostatin LAR depot)Pasireotide LAR (Signifor LAR) |
Inhibits GH secretion by binding to somatostatin 1,2,3 and 5 receptors | IM injection monthly IM injection monthly |
Diarrhea Biliary sludge/stonesHyperglycemia |
Hyperglycemia Pain at injection siteCardiac effects, gall bladder disease |
|||
| Lanreotide autogel (Somatuline depot) |
Inhibits GH secretion by binding to somatostatin 2 and 5 receptors | Deep SC injection monthly | Diarrhea Biliary sludge/stones |
Hyperglycemia | |||
| GH receptor antagonist | Pegvisomant (Somavert) | Binds to GH receptor but prevents dimerization of the receptor, thereby preventing receptor signaling, which results in decreased IGF-1 level | 6 days | SC injection daily | Transaminitis | Growth of the GH-secreting adenoma | |
Corticotroph (ACTH) Adenoma
Excess secretion of ACTH results in Cushing’s syndrome (Cushing’s disease if due to a pituitary adenoma) as a result of excessive stimulation of the adrenal cortex and hypersecretion of cortisol. ACTH is normally under negative feedback inhibition by circulating levels of cortisol as shown in Figure 4.
| Figure 4. Physiology of ACTH Secretion | |
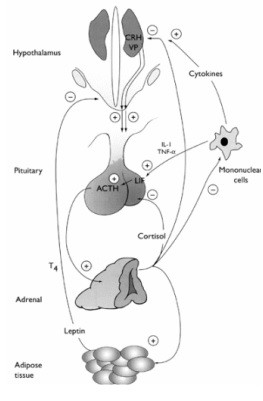 |
CRH stimulates ACTH secretion by activating protein kinase A after binding to CRH receptors on the corticotrophs. Vasopressin is a weak ACTH-releasing factor but acts together with CRH to potentate ACTH secretion. This synergistic action may be important during times of stress. ACTH stimulates cortisol secretion from the adrenal cortex. Cortisol then feeds back on corticotrophs in the pituitary to inhibit the biosynthesis and secretion of proopiomelanocortin (POMC), the precursor of ACTH, and inhibits the biosynthesis and secretion of CRH and vasopressin. Leptin, a fat-derived protein, and cytokines may also influence CRH secretion. |
ACTH-secreting pituitary adenomas are partially autonomous such that they retain feedback inhibition by cortisol but at a higher set point. Hence, ACTH and cortisol can be suppressed in individuals with ACTH-secreting pituitary adenomas if given a large enough dose of glucocorticoids.
Classically, a semi-synthetic steroid such as dexamethasone was used to assess suppressibility because it does not interfere with radioimmunoassays that measure cortisol.
By administering graded doses of dexamethasone (Liddle test) to individuals suspected of having Cushing’s syndrome and measuring urinary free cortisol levels, it is possible in most instances to separate out which individuals have adrenal adenomas, ACTH-secreting adenomas, or secrete ACTH ectopically (eg. oat cell carcinoma, medullary thyroid carcinoma, islet cell tumors, pheochromocytoma), from individuals who are simply obese. Normal (or obese) individuals will suppress urinary free cortisol levels to less than 20 µg/dl with low dose dexamethasone (0.5 mg every 6 h for 2 days), whereas in general, individuals with Cushing’s for any reason will not. Furthermore, individuals with an ACTH-secreting pituitary adenoma will suppress urinary free cortisol to >70-80% of baseline with high dose dexamethasone (2.0 mg every 6 h for 2 days), whereas this does not generally occur with the ectopic ACTH syndrome or adrenal adenomas. Adrenal adenomas can be separated from ectopic ACTH syndrome by measuring a plasma ACTH level. This tends to be very low in adrenal adenomas due to suppression of anterior pituitary corticotrophs by autonomous secretion of cortisol from the adrenal adenoma, and very high in the ectopic ACTH syndrome.
Unfortunately, it is now realized that there can be a considerable degree of overlap between ACTH-producing adenomas, and ACTH secreted ectopically from carcinoid tumors, making the distinction difficult between these two entities using the high dose dexamethasone suppression test. Nevertheless, the dexamethasone suppression test is still valuable in differentiating normal or obese individuals from those with Cushing’s. A simplified test that involves the administration of only 1 mg of Dexamethasone at 11PM and the measurement of an 8AM serum cortisol the following morning (overnight dexamethasone suppression test) has largely replaced the Liddle test. In most instances, normal individuals will suppress serum cortisol to <5 μg/dl, and often to <1.8 μg/dl due to the exquisite sensitivity of normal anterior pituitary corticotrophs to dexamethasone.
Alternatively, measurement of a late night plasma or salivary cortisol can be used to distinguish individuals with Cushing’s from normal individuals. This is based on the normal physiologic diurnal variation of cortisol, which peaks at around 8 AM and has its nadir at around midnight. Thus, a normally suppressed cortisol at midnight excludes the diagnosis of Cushing’s. This test is particularly useful to distinguish individuals that may have clinical and biochemical features that suggest Cushing’s disease but do not actually have Cushing’s disease. These individuals are said to have pseudo-Cushing’s (also known as non-neoplastic or physiologic hypercortisolism). Pseudo-Cushing’s can be caused by excessive alcohol intake, depression, severe obesity, and severe illness and may be due to alterations in the set point for feedback inhibition of CRH or ACTH by cortisol. The normal circadian rhythm of cortisol is preserved in pseudo-Cushing’s, which can be helpful to distinguish these individuals from true Cushing’s by measuring salivary cortisol levels at 8 AM and 11 PM. The abnormal dexamethasone suppression reverts to normal upon correction of the precipitating cause.
The following schema can be used to establish the diagnosis of Cushing’s and to identify the major subtypes:
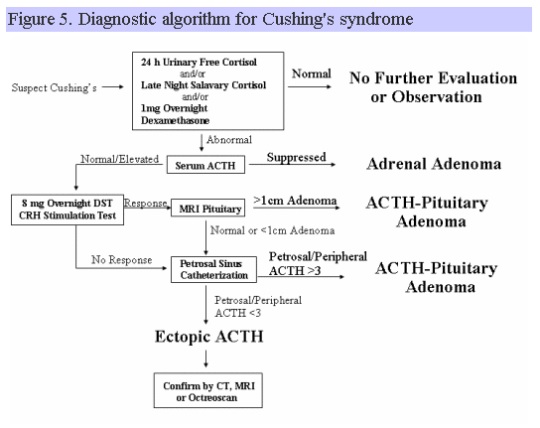 Individuals with Cushing’s disease may have a variety of clinical manifestations, depending upon the severity and duration of the disorder before diagnosis. A number of characteristic features are listed in Table 7.
Individuals with Cushing’s disease may have a variety of clinical manifestations, depending upon the severity and duration of the disorder before diagnosis. A number of characteristic features are listed in Table 7.
Table 7. Features of Cushing’s Disease
| General | Cardiovascular | Psychiatric |
| General Obesity | Hypertension | Depression |
| Cutaneous | Musculoskeletal | Metabolic |
| Facial Plethora Hirsutism Wide Striae (>1cm) Bruising Supraclavicular Fullness |
Proximal Myopathy Osteopenia Osteoporosis Back Pain Muscle Wasting |
Glucose Intolerance Diabetes Mellitus Hypokalemia Hypercalcuria |
Treatment of Cushing’s disease is primarily surgical, removal of the pituitary adenoma by transsphenoidal surgery. For individuals not cured by transsphenoidal surgery, conventional radiation or gamma knife therapy, bilateral adrenalectomy and/or medical therapy (shown in table 8) are options.
| Table 8. Medical Therapy for Cushing’s Disease | |
| Drug | Mechanism of Action |
| Ketoconazole
Levoketoconazole |
Antifungal agent; blocks cholesterol side-chain cleavage to reduce cortisol. May have independent inhibitory actions directly on ACTH secretion. |
| Metyrapone
Osilodrostat |
Inhibits 11β -hydroxylase to reduce cortisol. May also cause hypertension and hypokalemia due to increase in 11-deoxycorticosterone, and hirsutism due to increased androgens. |
| Aminoglutethimide | Often used together with metyrapone to reduce side effects. Inhibits cholesterol side-chain cleavage and 11β /18-hydroxylation. May cause hypothyroidism by interfering with iodine incorporation into thyroid hormone. |
| Mitotane | Destructive to adrenal cortex. Frequent adverse reactions. May cause hypercholesterolemia due to activation of HMG coenzyme reductase. |
| Mifepristone (RU 486) | Glucocorticoid antagonist. Approved in the US in 2012 for hyperglycemia related to Cushing’s disease. |
| Pasireotide | Somatostatin analog. Binds somatostatin receptor subtypes 1,2,3, and 5. Approved in the US in late 2012. |
| Etomidate | Hypnotic facilitating GABA neurotransmission. Rapidly reduces cortisol by blocking 11 hydroxylase. Must be given intravenously and in an intensive care unit setting. |
ThyrotrOph (TSH) Adenomas
Thyrotroph adenomas are very rare pituitary adenomas. The vast majority of these tumors are macroadenomas and, therefore, may be associated with mass effects. The clinical presentation is that of thyrotoxicosis due to excessive TSH secretion. A goiter occurs in greater than 90% of cases. Graves’ disease is often misdiagnosed in these patients, but the inappropriate increase in TSH for the elevated levels of thyroid hormones (T4 and T3) and the absence of ophthalmopathy, allow differentiation between these two disorders. Resistance to thyroid hormone due to mutations of the thyroid hormone receptors may also result in seemingly inappropriately elevated TSH levels for the circulating levels of thyroid hormones, but can be differentiated from thyrotroph adenomas by the high serum, α-subunit to TSH molar ratio found with thyrotroph adenomas (alpha-subunit [ng/ml] / TSH [mIU/ml] >1 in TSH adenomas).
Transsphenoidal surgery is the primary therapy for thyrotroph adenomas, but complete removal is difficult because of the large size of these tumors. Radiation therapy and/or medical therapy with octreotide are used for individuals with persistent disease. Octreotide is very effective in reducing TSH secretion to normal and reversing thyrotoxicosis, but is not very effective in shrinking the tumor.
Non-Secreting Adenomas
The majority of nonsecreting adenomas (90%) are actually glycoprotein hormone-producing adenomas, and contain various subunits of the glycoprotein hormones including α-subunit, β-LH and β-FSH. Some of these tumors actually do secrete FSH or α-subunits, but because these subunits are clinically nonfunctional, syndromes such as described above for acromegaly or Cushing’s disease, are not usually observed. A small number of males with hypersecretion of intact FSH have been described and had testicular enlargement. As a result, most of these tumors come to attention because of mass effects and/or hypopituitarism. Hyperprolactinemia is usually present due to stalk compression by the large mass and needs to be differentiated from a prolactinoma.
Treatment depends upon the size of the adenoma. Small adenomas do not necessarily require treatment if they do not compress local structures or cause hypopituitarism. Periodic MR images to assess the rate of growth may be all that is necessary. Large tumors are treated by transsphenoidal surgery. Complete resection of the tumor is not necessary. Relief of compressive symptoms is adequate. If hypopituitarism persists following surgery or is caused by the surgery, replacement therapy is given. Occasionally, anterior pituitary function may recover after decompression of the normal residual anterior pituitary.
If the adenoma grows following surgery, radiation therapy can be given. While many of these tumors do have somatostatin receptors, they respond poorly to medical treatment with octreotide and several studies have shown no shrinkage with this drug. Some nonsecreting adenomas also contain dopamine receptors, but their response to dopamine agonists such as bromocriptine and cabergoline are poor. Because nonsecreting adenomas are primarily gonadotroph adenomas and contain GnRH receptors, attempts to shrink the tumor with GnRH analogues (agonists and antagonists) have been tried but also without success.
Pathology
Tumors
Primary tumors of the pituitary constitute about 10 % of intracranial tumors. About two-thirds of pituitary tumors are adenomas arising in the adenohypophysis. The second commonest type of pituitary tumor is the craniopharyngioma, which is believed to arise from remnants of Rathke’s pouch and occurs about one‑fourth as often as adenomas. Other tumors are rare. They include angiomas and meningiomas arising from blood vessels and meninges; granular cell tumors and gliomas from the neurohypophysis, and germinomas and teratomas from germ cell rests formed early in embryogenesis.
“Incidental” microscopic pituitary adenomas and granular cell tumors are found in a high percentage of the population. As with other incidental endocrine tumors, there is currently no way to predict which tumors would have gone on to be clinically significant.
As might be expected from their origins and anatomic location, pituitary tumors may produce clinical symptomatology in two general ways: (1) by direct production of hormones resulting in endocrine hyperfunction; and (2) by expansion, resulting in hypopituitarism from destruction of normal cells or interference with transport of hypothalamic hormones; and in neurological signs, such as visual defects from encroachment on the optic chiasm. The first endocrine symptoms, usually amenorrhea and loss of libido, often precede visual signs by long periods, sometimes decades. The only pituitary tumors that directly cause endocrine hyperfunction are hormone-producing tumors of the adenohypophysis. However, endocrinologically inactive tumors can elevate serum prolactin levels indirectly by interfering with hypothalamic-hypophyseal transport of dopamine. These elevations are generally lower than those produced directly by functional tumors.
Malignant pituitary tumors, such as carcinomas of the adenohypophysis or malignant craniopharyngiomas, are extremely rare if malignancy is defined, as in most other organs, by capacity to metastasize. Some authors define malignancy in pituitary tumors as capacity to invade, rather than compress, adjacent structures. This phenomenon occurs in a gross clinically significant fashion in about 3% of pituitary tumors. Microscopic foci of invasion, however, occur commonly.
Adenomas
Pathology Nomenclature and Function.
Pituitary adenomas are composed of a diffuse, monotonous population of cells that lacks the nested, 3-color pattern of the normal gland in H&E sections. The classification of pituitary adenomas is based primarily on the hormones produced, defined principally by immunohistochemistry. Additional characteristics including expression of transcription factors and cytoskeletal filaments define subclassifications that are prognostically useful and can help to guide therapy. Distinctive ultrastructural features (for example, “densely granulated” or “sparsely granulated” somatotroph adenomas which would correspond respectively to acidophil or chromophobe adenoma in H&E stains), are sometimes also considered. Examples of these subclassifications are in the table below.
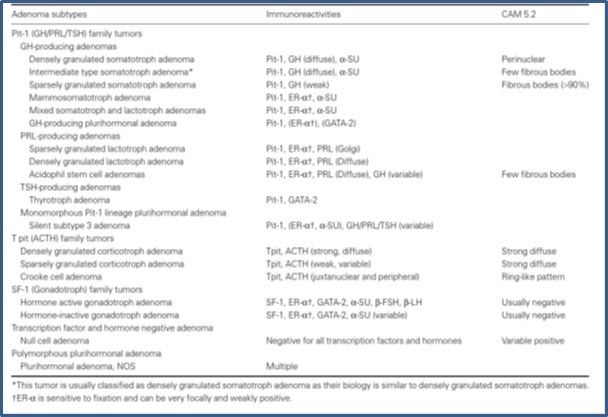
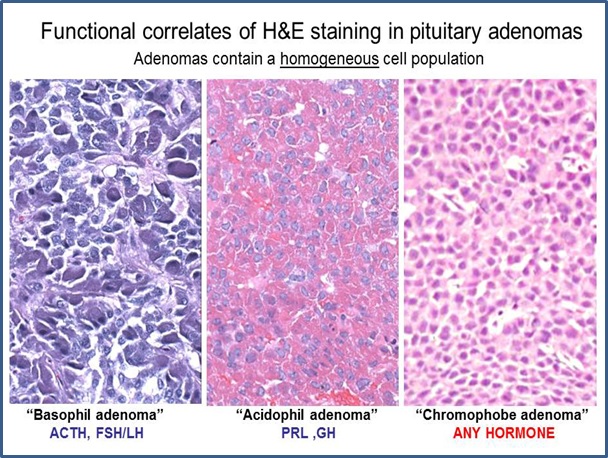
Concepts regarding pituitary adenomas evolved in parallel with understanding of the normal gland. Like the normal adenohypophysis, pituitary adenomas were historically defined on a three‑color system, as acidophilic, basophilic, or chromophobic. Although H&E staining is no longer the basis for tumor classification, it is still a useful pathology tool for pattern recognition. Because tissue samples in trans-sphenoidal surgery are often small, the first task for the pathologist is to distinguish pituitary adenoma from normal or hyperplastic pituitary. H&E staining is the usual first approach, especially for intraoperative frozen sections that require rapid assessment of tissue sampling. In routine H&E staining , about two‑thirds of adenomas are chromophobic, slightly less than one‑third are acidophilic, and the remainder (less than 5%) are basophilic. Staining reflects a balance between the ability to synthesize a hormone and ability to store the hormone in secretory granules. Chromophobic staining simply reflects sparse granule content and, therefore, chromophobic tumors can produce any hormone or none and can be clinically functional or clinically silent. Acidophilic and basophilic adenomas usually produce the same hormones as their normal cellular counterparts. They can also be clinically functional or clinically silent depending on how much hormone they secrete.
Both the therapy and the prognosis of pituitary adenomas are influenced by a tumor’s hormonal profile. The majority of pituitary adenomas produce prolactin. GH is the second most common hormone. Most of tumors produce multiple hormones, usually GH plus prolactin. This combination may reflect the presence of dual function acidophils in the normal pituitary. Clinical syndromes, however, are usually chiefly attributable to a single hormone. Gonadotroph adenomas were until fairly recently considered uncommon, but are now recognized to account for a large percentage of tumors that clinically appear to be inactive. Thyrotrophic tumors are rare.
It is important to remember that although the great majority of pituitary adenomas are biochemically functional, most are clinically silent, particularly if microadenomas are considered. In autopsy series, more than 25% of apparently normal individuals are found to have microadenomas. Most lesions in such patients are prolactinomas or “null cell” adenomas. Microadenomas can clinically be either functional or non-functional, depending on the patient’s sex. Studies of serum prolactin levels in secondary amenorrhea have resulted in detection of many prolactin‑producing microadenomas in females, while the same lesions in males go undetected. Microadenomas have also been reported to be present in most cases of Cushing’s disease in which the sella is radiologically normal. Immunohistochemical profiling of the hormones produced by clinically silent adenomas can provide clinically important information. For example, silent corticotroph adenomas are more aggressive and more prone to recurrence than corticotroph adenomas associated with Cushing’s disease.
Etiology, Regulation and Potential Targeted Therapies
Several germline and somatic mutations lead to development of pituitary adenomas. The former include inactivating mutations of the MEN1 tumor suppressor gene, and the latter include activating mutations of genes encoding G-protein coupled receptors that stimulate cyclic AMP production. A number of hypothalamic hypophysiotropic hormones utilize cyclic AMP as a second messenger, suggesting that these mutations cause tumors by mimicking the effects of hypothalamic hormones and activating the same signal transduction pathways that stimulate hormone secretion. The extent to which physiological levels of activation contribute to tumor development is unclear. Animal models support a role for hypothalamic hormones in the development of some types of pituitary tumors (e.g., TSH adenomas arising after thyroidectomy). In humans, patients with pancreatic endocrine tumors producing ectopic GHRH can develop either pituitary adenomas or hyperplasias; and patients with hypothalamic Cushing’s disease can develop ACTH adenomas after bilateral adrenalectomy (Nelson’s syndrome).
Although mechanisms of causality are not completely established, it is clear that hormone production, cell proliferation and survival can be regulated in pituitary adenomas by agents involved in normal function, including estrogen, dopamine and somatostatin. In addition to hormonal profiling, pathologists will be increasingly called on to help exploit this type of regulation by identifying specific targets for therapy. These might include estrogen receptors, dopamine receptors that predict response to bromocriptine or other dopamine agonists, and somatostatin receptors that predict response to octreotide or pasireotide or other somatostatin analogs.
Pituitary Carcinomas
Pituitary carcinomas represent less than 0.2% of surgically treated pituitary tumors. As with several other types of endocrine tumors, there are no reliable histological features for diagnosis of malignancy. Malignancy is diagnosed by the presence of metastases, which may present several years after the initial surgery. Metastases may involve the subarachnoid space or distant organs, spread mostly by hematogenous routes.
Craniopharyngiomas
In contrast to pituitary adenomas, which for the most part occur in adults and are intrasellar, craniopharyngiomas are more commonly suprasellar than intrasellar, and are most common in children. They may, however, occur in adults as well. A bimodal distribution curve shows peak incidences at ages 5-20 years, and in the sixth decade. The patients may present with dwarfism, sexual dysfunction, or vasopressin deficiency (previously known as diabetes insipidus). The tumors are usually encapsulated and cause symptoms by compression of surrounding structures.
Histologically, craniopharyngiomas are readily diagnosed on routine H&E sections. Two histological types, adamantinomatous and papillary are described. The former, representing about 2/3 of cases, are often calcified, permitting diagnosis by x‑ray. Their name is derived from the fact that they may histologically resemble the enamel organ of developing teeth and odontogenic tumors of the jaw (enamel = adamantin, from Greek adamas, unyielding) possibly reflecting their oral ectodermal ancestry.Typically they contain admixtures of columnar and squamous epithelium, resembling the Rathke’s pouch remnants that are frequently observed as incidental findings, and have multiple cystic areas containing keratin, chronic inflammatory cells, cholesterol crystals and other debris. Although the tumors grow slowly, they are difficult to remove because of their location, and their long‑term prognosis is poor. Papillary craniopharyngiomas have a somewhat better prognosis.
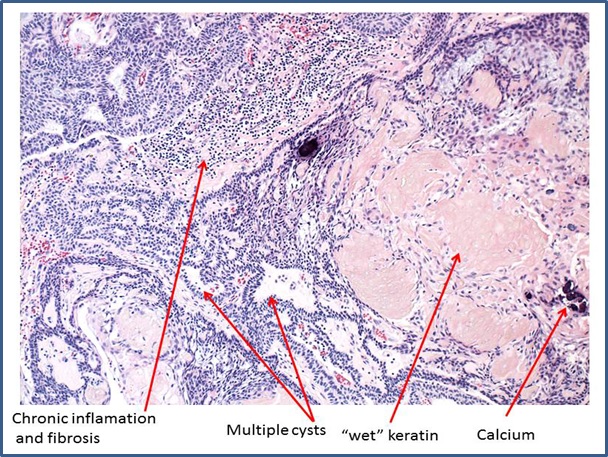
Histologically, craniopharyngionmas are readily diagnosed on routine H&E sections. Two histological types, adamantinomatous and papillary are described. The former, representing about 2/3 of cases, are often calcified, permitting diagnosis by x‑ray. Their name is derived from the fact that they may histologically resemble the enamel organ of developing teeth and odontogenic tumors of the jaw (enamel = adamantin, from Greek adamas, unyielding) possibly reflecting their oral ectodermal ancestry.Typically they contain admixtures of columnar and squamous epithelium, resembling the Rathke’s pouch remnants that are frequently observed as incidental findings, and have multiple cystic areas containing keratin, chronic inflamatory cells, cholesterol crystals and other debris. Although the tumors grow slowly, they are difficult to remove because of their location, and their long‑term prognosis is poor. Papillary craniopharyngiomas have a somewhat better prognosis.
Feedback/Errata