
3 Pituitary Insufficiency and Pathology
Ronald M. Lechan, MD, PhD and Arthur Tischler, MD
Learning Objectives
- To list several causes of pituitary insufficiency
- To describe symptoms of pituitary insufficiency due to deficiencies in specific hormones
- To differentiate between basal and dynamic testing in hypopituitarism
- To discuss lab tests for the various hypohormonal states of pituitary insufficiency
- To name medications used in the treatment of hypopituitarism and how they should be monitored
- Describe the major ways that the embryological origins of the pituitary are reflected in pituitary pathology.
- Describe the major causes of pituitary hypofunction and recognize the major pathological findings.
Key Concepts
- Pituitary insufficiency (hypopituitarism) may arise from congenital defects or acquired disease of the pituitary or hypothalamus.
- Pituitary hormone levels may be inappropriately normal relative to target hormone levels in hypopituitarism. For example, a normal TSH may be seen with a low free T4 in a patient with untreated central hypothyroidism.
- To test ACTH reserve directly, an insulin tolerance test can be done (also tests GH reserve). Metyrapone, which inhibits the last step in cortisol synthesis, can also test ACTH reserve.
- A dehydration test can be done to assess for central diabetes insipidus where ADH will be low with a high serum osmolality and low urine osmolality.
- The presence of diabetes insipidus, slightly elevated prolactin and one or more anterior pituitary deficiencies suggests involvement of the pituitary stalk or hypothalamus.
- Glucocorticoids must be replaced first if multiple hormones are deficient.
- Normal adenohypophysis contains a heterogeneous, nested population of neuroendocrine-type secretory cells that on H&E stain are acidophilic (cytoplasmic granules stain with eosin, an acidic dye; make GH and Prolactin), basophilic (cytoplasmic granules stain with hematoxylin, a basic dye; make ACTH, FSH/LH), or chromophobic (make any hormone, are sparsely granulated).
- Neurohypophysis consists of axons and glia, no secretory cell bodies
- The adenohypophysis has a large functional reserve. Small asymptomatic infarcts occur in various conditions (sickle cell disease, hypovolemic shock etc.). Large infarcts causing functional impairment are most common in obstetric shock (Sheehan’s syndrome).
- Pituitary apoplexy is an infarcted pituitary adenoma.
- Common cancers, e.g. breast and lung, frequently metastasize to the pituitary and can selectively affect the neurohypophysis, causing diabetes insipidus.
- Empty sella syndrome is an intrasellar arachnocoele
- Differential dx of intrasellar cysts includes Rathke cleft cyst (squamous, cuboidal, columnar, sometimes respiratory lining, sometimes with goblet cells), dermoid cyst (looks like complete skin with dermal appendages) epidermoid cyst (looks like epidermis without dermal appendages).
Definition
Hypopituitarism is a term that refers to the deficiency of one or more anterior and/or posterior pituitary hormones. The deficiency may be total (panhypopituitarism) or partial, in which one or more of the pituitary hormones may be deficient.
Hypopituitarism
Etiology of Hypopituitarism
There are many disorders that can cause hypopituitarism, either by affecting the hypothalamus or the pituitary gland. Congenital disorders are a cause for hypopituitarism presenting in children. Formation of the pituitary during embryonic development depends upon the juxtaposition of cells of neurectodermal origin, which form the posterior pituitary, and endodermal cells which form the anterior pituitary. Defects in the transcription factors HESX-1, PROP-1, and PIT-1 are known to result in various degrees of hypopituitarism. HESX-1 mutations are associated with septo-optic dysplasia, characterized by the triad of optic nerve hypoplasia, midline neuroradiological abnormalities such as agenesis of the corpus callosum, and pituitary hypoplasia with hypopituitarism. PROP-1 mutations have deficiencies in the gonadotrophs (LH and FSH), growth hormone, prolactin and TSH. PIT-1 mutations result in combined deficiencies of growth hormone, prolactin and TSH. Selective deficiencies in LH and FSH may arise from mutations in the KAL gene that encodes anosmin, the fibroblast growth factor receptor 1 (FGFR1), fibroblast growth factor 8 (FGF8), prokineticin 2 (PRO2) and prokinecticin receptor 2 (PROR2), all interfering with the ability of hypothalamic neurons that regulate gonadotropin secretion to establish their proper position in the hypothalamus and commonly associated with a loss of sense of smell (anosmia). Mutations may also selectively affect posterior pituitary function including point mutations in the vasopressin-neurophysin II gene. Most involve the neurophysin II region of the vasopressin gene, resulting in abnormal processing, transport or cleavage of the vasopressin precursor, or cytotoxicity to vasospressin-producing cells in the hypothalamus. Wolfram syndrome (DIDMOAD syndrome) is an inherited autosomal recessive disorder due to a mutation in the transmembrane protein, wolframin, and characterized by DI, Diabetes Mellitus, Optic Atrophy and Deafness. Pituitary adenomas are the most common cause for hypopituitarism in adults. Causes of hypopituitarism are listed in Table 1.
| TABLE 1. Causes of Hypopituitarism. | ||
| Congenital Disorders | ||
| HESX-1, PIT-1, and PROP-1 mutations, KAL, FGFR1, FGF8, PRO2, PROR2 and DAX-1 mutations, GnRH and GHRH receptor mutations, Kallmann, PraderWilli and Bardet-Beidl Syndromes, Vasopressin-Neurophysin gene mutations, DIDMOAD | ||
| Benign Neoplasms | Malignant Neoplasms | Cysts |
| Pituitary Adenoma Craniopharyngioma Meningioma Optic Nerve Glioma |
Germ Cell Tumors (Germinoma) Lymphoma Plasmacytoma Metastatic Disease (breast, lung) |
Arachnoid Dermoid Epidermoid Rathke’s Cleft |
| Granulomatous Disease | Vascular Disorders | Infections |
| Eosinophilic Granuloma Histiocytosis X Sarcoidosis |
Aneurysm Cavernous Angioma Infarction (Postpartum, D.M.) |
Abscess Cysticercosis Tuberculosis |
| Autoimmune | Traumatic | Other |
| Lymphocytic Hypophysitis Vasculitis |
Pituitary Stalk Transection | Cerebral Edema Pituitary Apoplexy Radiation Therapy |
Clinical Presentation of Hypopituitarism
Disorders that affect the hypothalamus may be distinguished from those affecting the pituitary by using both clinical and biochemical parameters. Since prolactin secretion from the anterior pituitary is primarily under inhibitory regulation by the hypothalamus, any destructive process of the hypothalamus often results in elevation of prolactin levels in the bloodstream (hyperprolactinemia). The combination of a modestly elevated prolactin and deficiency of one or more anterior pituitary hormones, therefore, suggests involvement of either the hypothalamus or pituitary stalk. In contrast, destruction of the anterior pituitary would be expected to result in low prolactin levels. An exception to this rule, however, occurs when prolactin is abnormally secreted from a prolactinoma. In addition, disorders affecting the hypothalamus are more commonly associated with abnormalities of posterior pituitary function such as diabetes insipidus (see below), and a variety of other hypothalamic syndromes listed in Table 2. A lesion centered in the median eminence due to metastatic breast carcinoma, for example, might be expected to result in deficiencies of ACTH, TSH, GH, LH/FSH, and vasopressin, and a modest elevation in prolactin. Destruction of the anterior pituitary, such as may occur in the postpartum period due to bleeding into the anterior pituitary (Sheehan’s Syndrome), might result in deficiencies of ACTH, TSH, GH, LH/FSH, as well as prolactin, but usually not vasopressin.
| TABLE 2. Neurologic Manifestations of Hypothalamic Disease. | |
| Disorders of Temperature Regulation | Disorders of Food Intake |
| Hyperthermia Hypothermia Poikilothermia |
Anorexia Hyperphasia |
| Disorders of Water Intake | Disorders of Sleep and Consciousness |
| Adipsia, Hypodipsia Compulsive Water Drinking |
Somnolence Coma |
| Disorders of Autonomic Function | Disorders of Psychic Function |
| Cardiac Arrhythmias Diencephalic Epilepsy Pulmonary Edema |
Rage Hallucinations |
Since the pituitary gland lies adjacent to the cavernous sinus, temporal lobes and sphenoid sinus (Fig. 5), other clinical symptoms such as palsies of extraoccular muscles, sensory disturbances in the first and second branches of cranial nerve V, temporal lobe seizures, and CSF rhinorrhea can occur with disorders affecting the pituitary gland.
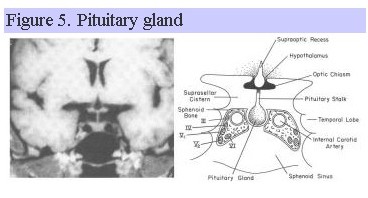
Disorders of both the hypothalamus and pituitary can also compress or infiltrate the optic chiasm, resulting in a variety of visual field abnormalities affecting peripheral vision (hemianopsia). Specific symptoms associated with the loss of each pituitary hormone are listed in Table 3 and more fully discussed below. GH is often the first hormone to be affected during hypopituitarism, followed by LH/FSH, ACTH, TSH and prolactin. Nevertheless, any of the anterior pituitary hormones can be affected in any order. In childhood, the dominant clinical picture of hypopituitarism is often failure of normal linear growth, while in the adult it is hypogonadism. Depending upon the underlying cause, however, pituitary deficiencies may be gradual and progressive (as with an enlarging pituitary adenoma) or acute (as with pituitary apoplexy). As a result, the presenting manifestations of hypopituitarism can vary enormously.
| TABLE 3. Symptoms Associated with Hypopituitarism Due to Deficiency of Specific Hormones | |
| Growth Hormone (GH) | Gonadotrophins (LH/FSH) |
| Weight Gain (abdominal adiposity) Decreased Muscle Strength Decreased Exercise Capacity Increased Cardiovascular Risk Impaired Psychological Well-being Growth Retardation (children) |
Amenorrhea/oligomenorrhea Infertility Dyspareunia Breast Atrophy Loss of Secondary Sexual Hair Decreased Libido Impotence Small, Soft Testes Decreased Muscle Mass and Strength Decreased Erythropoiesis Osteoporosis |
| Thyroid Stimulating Hormone (TSH) | Adrenocorticotropin (ACTH) |
| Sensitivity to Cold Dry Skin Constipation Decreased Energy |
Weight Loss Fatigue Pallor Hypoglycemia Nausea/Vomiting Circulatory Collapse |
| Prolactin | Vasopressin (Antidiuretic Hormone) |
| Poor or Absent Lactation | Urinary Frequency Thirst |
Biochemical Diagnosis of Hypopituitarism
Although one can infer some pituitary deficiencies based on history and clinical findings, demonstrating hormone deficiencies by biochemical testing is essential to establish the diagnosis of hypopituitarism. A variety of tests can be performed to evaluate the reserve of each of the pituitary hormones and are described below under each hormone. It is extremely important to make the diagnosis of ACTH and/or TSH deficiency, since these anterior pituitary hormones are essential for life. Some tests are also helpful in differentiating whether the cause for hypopituitarism is of pituitary or hypothalamic origin.
Hypocortisolism (ACTH)
Cortisol and adrenal androgen secretion are ACTH dependent and can be decreased in hypopituitarism. Because aldosterone secretion is dependent primarily on the renin-angiotensin system, however, hypopituitarism is not associated with aldosterone deficiency. Major symptoms of ACTH deficiency include fatigue, weakness, anorexia, weight loss, nausea, vomiting, abdominal pain, and myalgia. Pallor of the skin and decreased tanning are observed contrary to the characteristic manifestations of Addison’s Disease due to autoimmune adrenalitis. Because of impaired gluconeogenesis, hypoglycemia may be present particularly with fasting or alcohol ingestion, and hyponatremia is common due to inappropriate secretion of vasopressin (SIADH) and inability to secrete a water load. Loss of adrenal androgens is of little consequence for males if testicular androgen production is normal, but can result in decreased libido and loss of axillary and pubic hair in females. Circulatory collapse can occur in association with acute stressful situations. Several tests are available to assess ACTH reserve. Measurement of basal levels of serum cortisol are generally not very helpful except when extremely low since cortisol does undergo a diurnal variation (higher in the AM and lower in the PM) and does not necessarily reflect adequate reserves of ACTH required during stress. Thus, provocative tests of the adrenal axis are necessary.
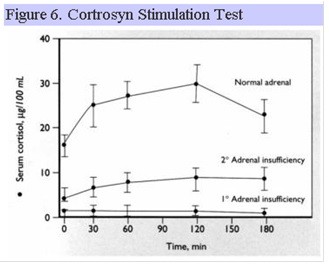
Cortrosyn Stimulation Test
Cortrosyn is synthetic ACTH 1-24 and is administered IM or IV to induce cortisol secretion from the adrenal gland. Individuals with long-standing hypopituitarism develop atrophy of the adrenal cortex and as a result, show blunted peak cortisol responses to cortrosyn (Fig. 6).
Overnight Metyrapone Test
Metyrapone inhibits 11-beta hydroxylase in the adrenal cortex, thereby reducing the conversion of 11-deoxycortisol to cortisol. The reduction in cortisol in the circulation increases ACTH secretion through direct effects on anterior pituitary corticotrophs (negative feedback regulation). This results in a secondary increase in 11-deoxycortisol (compound S) in the circulation. ACTH and 11-deoxycortisol are measured in the blood the morning following a single dose of metyrapone the preceding evening. In hypopituitarism, both ACTH and 11-deoxycortisol levels are blunted.
Insulin Tolerance Test
The insulin tolerance test (ITT) is the “gold standard” for assessing adequate adrenal reserve. However, the test is potentially hazardous since hypoglycemia (blood glucose less than 40 mg/dl) is a necessary endpoint to allow activation of the adrenal axis. Accordingly, the ITT is contraindicated in anyone with a history of heart disease or seizures and should be avoided in the elderly. The stress of the hypoglycemia increases the secretion of ACTH through effects on the release of CRH and vasopressin from the hypothalamus. Both ACTH and cortisol are measured after achieving hypoglycemia. In hypopituitarism, both ACTH and cortisol levels are blunted.
Hypothyroidism (TSH)
TSH insufficiency results in reduced thyroid hormone secretion from the thyroid gland resulting in symptoms of hypothyroidism. Classical features include cold intolerance, fatigue, dry skin and constipation. Speech may be slowed, and there may be slowing of mental and physical function. Hyponatremia and a normochromic normocytic anemia may be present. As opposed to ACTH, the best test to assess TSH reserve is measurement of basal thyroid hormone levels. Low thyroxine (T4) and triiodothyronine (T3) levels simultaneously with an inappropriately low TSH, establishes the diagnosis of central hypothyroidism.
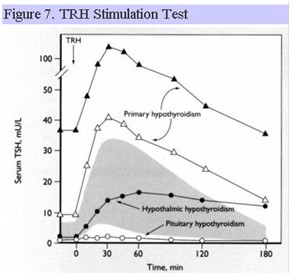
TRH Stimulation Test
Synthetic TRH can be administered IV and acts directly on the anterior pituitary. In normal individuals, TSH rises abruptly (within 15-30 min) and then falls to baseline values. Pituitary disease may result in an absent or blunted TSH response, whereas hypothalamic disease may result in a delayed peak response of TSH (Fig. 7). Significant overlap exists in the TSH response to TRH in patients with disorders that affect the pituitary and hypothalamus, respectively, such that this test cannot reliably differentiate between pituitary and hypothalamic disease. TRH is no longer clinically available for diagnostic testing in the US.
 Prolactin
Prolactin
The only clinical manifestation of prolactin deficiency known is failure of lactation in the postpartum period. Basal levels of prolactin are generally sufficient to assess prolactin reserve. Serum prolactin may be low with pituitary lesions (unless due to a prolactinoma) and high with hypothalamic disorders or disorders that prevent the flow of portal blood from the hypothalamus to the anterior pituitary such as stalk compression. A number of provocative tests are available that are very effective in stimulating the secretion of prolactin, including the TRH stimulation test and the ITT, but are rarely needed.
Hypogonadism
LH and FSH are necessary for secondary sexual development, maintenance of secondary sexual characteristics and fertility. In males, gonadotrophin deficiency results in loss of libido, erectile dysfunction, oligospermia, reduced erythropoiesis, decreased body mass and osteoporosis. In females, LH and FSH deficiency are manifest by oligo/amenorrhea, breast atrophy, decreased secondary sexual hair and osteoporosis. Low plasma estradiol (females) or testosterone (males) levels simultaneous with inappropriately low LH and FSH levels, establishes the diagnosis of hypogonadotropic hypogonadism. Provocative tests are available including the GnRH stimulation test, in which synthetic GnRH acts directly on the anterior pituitary, and the clomiphene stimulation test, in which clomiphene acts on the hypothalamus as an anti-estrogen, but also are not very effective in differentiating pituitary from hypothalamic disease.
Growth Hormone Deficiency (GH)
Assessment of GH reserve is particularly important in children who have not yet reached their full growth potential. However, as it is becoming increasingly apparent that GH deficiency may be associated with significant physical and psychological abnormalities in adults and could contribute to the 2-3-fold increased mortality in individuals with hypopituitarism primarily due to cardiovascular complications, assessment of GH reserve in adults is also important. Adults with growth hormone deficiency exhibit a higher body mass index and waist-to-hip ratio due to an approximately 8% increase in subcutaneous and visceral adipose tissue and approximately 8% decrease in lean body mass. There is a tendency for increased atherothrombotic propensity due to higher plasminogen activator inhibitor 1 (PAI-1) activity and increased fibrinogen, and abnormal lipid profiles are also commonly seen including elevated total cholesterol, LDL cholesterol, ApoB, and triglycerides. Elevated serum insulin and impaired glucose metabolism may also contribute to the increased risk of cardiovascular disease. Since GH is secreted episodically (one pulse every 2-4 h), measurement of a random GH level is not useful. In addition, unless the insulin like growth factor-1 (IGF-1) level is extremely low (IGF-1 is produced primarily by the liver and mediates many of the actions of GH), it also is not a reliable index of GH reserve. Thus, provocative tests are necessary to establish normal GH reserve.
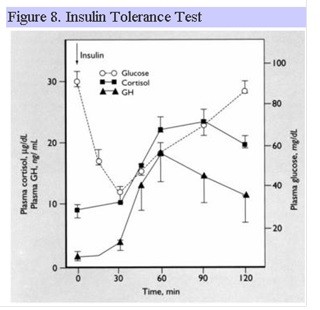
Insulin Tolerance Test
The ITT is the “gold standard” for assessing GH reserve. GH rises in response to the hypoglycemia induced by a bolus of insulin. Constant medical monitoring is required throughout the procedure as a blood glucose less than 40 mg/dl is required for an adequate GH response in normal individuals. The ITT can also be used to assess ACTH reserve as described above. Fig. 8 shows the normal response for GH and ACTH following insulin hypoglycemia.
 Arginine Stimulation Test
Arginine Stimulation Test
Arginine produces a rise in blood glucose, followed by a secondary fall. When the blood glucose falls, there is a rise in GH. Arginine is infused IV over 30 min and the GH response measured over 2 h. The test is not as reliable as the ITT.
GHRH Stimulation Test
Synthetic GHRH 1-29 is administered IV and the GH response measured over 2 h. Its action is exerted directly on the anterior pituitary. The biochemical diagnosis of GH deficiency is enhanced when the GHRH stimulation test is combined with the Arginine Stimulation Test.
Glucagon Stimulation Test
Glucagon is administered intramuscularly and the GH response measured over 3 h. Its mechanism of action is unknown but may be directly on the hypothalamus. GH peak tends to occur between 2-3 h. Can also be used to assess adrenal reserve.
MACImorelin Stimulation Test
Macimorelin is an orally active growth hormone secretagogue. It was approved for diagnostic testing in 2017. Testing is simple, generally well tolerated and comparable with the insulin tolerance test for the diagnosis of adult GH deficiency.
A number of other provocative stimuli are available for stimulating GH including clonidine, glucagon, L-DOPA, and exercise, but these tests are not as reliable in inducing a GH rise in normal individuals as the above tests, and therefore, are difficult to interpret.
ARginine Vasopressin DEficiency (also known as Central diabetes insipidus)
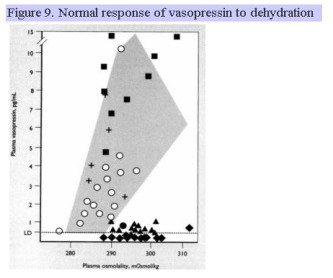
Clinical disease manifested by urinary frequency and large urine volumes (greater than 3 L/d but may reach 20 L/d) due to the inability to concentrate the urine is a condition referred to as arginine vasopressin deficiency (AVP-D). A similar clinical presentation may be arginine vasopressin resistance (AVP-R) is due to renal insensitivity to the actions of vasopressin. Characteristic of AVP-D is a high serum osmolality (usually 290-300 mOsm/kg), inappropriately low urine osmolality and specific gravity (usually <200 mOsm/kg with urine sp. gr > 1.010), and inappropriately low plasma vasopressin level. These individuals are generally very thirsty, unless they have simultaneous damage to thirst centers in the hypothalamus. If the plasma osmolality is not greater than 295 mosmol/kg, a water deprivation test is performed. The individual is deprived of all food and drink until the plasma osmolality rises above 295, at which time the urine osmolality and plasma vasopressin level are simultaneously measured. Individuals with AVP-D will have serum osmolality levels that are high, urine osmolality levels that are inappropriately low, and will continue to have a high urine output. The normal rise in plasma vasopressin with dehydration (rise in plasma osmolality) is shown in Fig. 9. The shaded area and + correspond to normal, expected values. Filled squares represent individuals with AVP-R, filled diamonds represent individuals with complete AVP-D, and closed triangles represent individuals with partial AVP-D. During a water deprivation test, no individual should be allowed to lose more than 3% of body weight. Following the administration of the synthetic vasopressin analogue, desmopressin or DDAVP, individuals with AVP-D show an increase in urine osmolality of >50% 60 min later. Copeptin is the C-terminal segment of the arginine vasopressin prohormone. It is another surrogate marker for vasopressin which is more stable and can be measured in the assessment of patients with polyuria. Similar to vasopressin, patients with central diabetes insipidus will have a low copeptin level after water deprivation.
 Management of Hypopituitarism
Management of Hypopituitarism
The aim of treatment is to replace those deficiencies needed for normal function and to treat the underlying disease process. In most instances, replacement of hormone deficiencies can be accomplished by the oral, cutaneous (skin patch, gels or creams), subcutaneous or nasal administration of the deficient hormones. The dose is usually the same from day to day with exception of glucocorticoid replacement, which must be increased at times of stress. Individuals with ACTH deficiency must wear a MedicAlert bracelet or necklace indicating their dependence on exogenous glucocorticoids. They should also be instructed in the administration of intramuscular glucocorticoids should they be unable to take their medication orally due to vomiting. Table 4 lists the common drugs used for hormone replacement therapy in hypopituitarism. As a general rule, glucocorticoid replacement therapy should be given first. Thyroid hormone replacement therapy should never be given before glucocorticoids have been replaced to prevent precipitating an Addisonian crisis.
| TABLE 4. Hormone Replacement Therapy in Hypopituitarism | |
| Deficient Hormone | Replacement |
| ACTH | glucocorticoids (hydrocortisone, prednisone, dexamethasone) |
| TSH | L-thyroxine (T4) |
| Prolactin | Not replaced |
| LH/FSH | estradiol + progesterone (females) testosterone (males) pulsatile synthetic GnRH (if anterior pituitary intact) or HCG/human recombinant FSH if fertility desired to induce ovulation/spermatogenesis |
| GH | human recombinant growth hormone (hGH) |
| Vasopressin (ADH) | desmopressin (DDAVP) |
Pathology
Developmental Abnormalities
- Anatomical malpositions of the pituitary lobes are exotically rare.
- Aplasia and hypoplasia of the adenohypophysis occasionally result from defective formation of Rathke’s pouch, causing varying degrees of hypopituitarism in infants. These conditions tend to occur in association with cyclopia or anencephaly, respectively, but may occur in the absence of other skull or brain abnormalities.
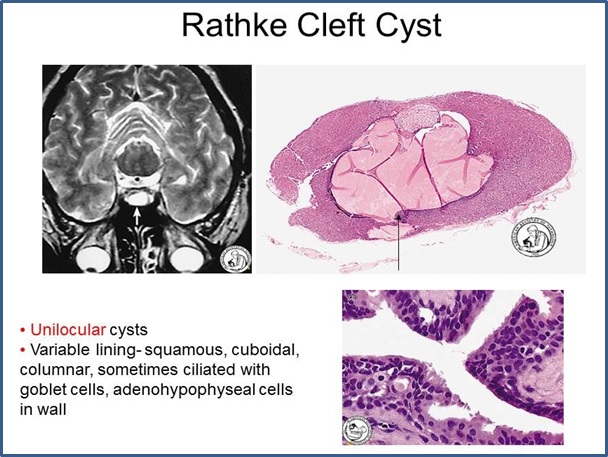
- Developmental remnants and cysts. Small nests of squamous cells reminiscent of Rathke’s pouch epithelium may occur in up to 30% of human pituitary glands. Originally believed to be direct remnants of Rathke’s pouch, it now appears that they arise from squamous metaplasia of adenohypophyseal endocrine cells. Colloid filled cysts, believed to be remnants of the lumen of Rathke’s pouch, are common findings between the anterior and posterior pituitary lobes, and usually are of no clinical significance. Occasionally cysts may expand enough to become symptomatic by virtue of their size. Such clinically significant “Rathke’s cleft cysts” are the most common type of cyst involving the pituitary but are very rare overall, accounting for only ∼3% of sellar masses. They are lined by cuboidal and columnar epithelium including ciliated cells and goblet cells. The type of epithelium distinguishes them from epidermoid cysts (resembling epidermis) and dermoid cysts (resembling complete skin including dermis and appendages), which are the major differential diagnoses.
Empty Sella Syndrome
Empty sella syndrome is an arachnocele resulting from a defect in the diaphragma sellae. The defect may be primary or may result from surgery, necrosis or infarction. Increased CSF pressure inside the sella compresses the pituitary and enlarges the sella so that it appears empty in imaging studies. However, pituitary function is often preserved.
Circulatory Disturbances
- Infarction: The functional reserve of the adenohypophysis is sufficiently great that hypopituitarism will not become apparent until about 70% of its tissue is destroyed. Small asymptomatic foci of infarction or small scars are commonly observed in autopsy pituitaries, particularly in association with diabetes mellitus, shock, cerebrovascular accident, sickle cell anemia, or other conditions producing impaired blood flow. The majority of cases of hypopituitarism occurring on a vascular basis result from obstetric shock related to postpartum hemorrhage, or Sheehan’s Syndrome. The pituitary appears to be more prone to necrosis in obstetric than in non-obstetric shock, and postpartum pituitary necrosis may destroy the adenohypophysis almost totally. The pathogenesis of this condition is not established, but Sheehan proposed that it results from arteriolar spasm in the vessels supplying the pituitary portal venous system. The neurohypophysis is generally, but not always, spared, because it is not dependent on the portal vessels for the majority of its blood supply.
- Hemorrhage: Hemorrhage in the pituitary occurs in more than half of patients dying from head trauma. Bleeding in these cases predominantly affects the neurohypophysis and the pituitary capsule and results from contusions and shearing forces. The adenohypophysis is more selectively affected by bleeding in pituitary tumors. An extreme form of the latter is “pituitary apoplexy” in which bleeding in a tumor may produce signs of acute expansion, followed by destruction of tumor and adjacent tissue and onset of hypopituitarism.
Involvement in Miscellaneous Diseases
A. Metastatic Carcinoma
Although primary malignancies of the pituitary are rare, metastases to the pituitary gland have been reported in l 3% of all cancer patients at autopsy. The commonest primary sites are first breast, and second, lung. The neuro-hypophysis is involved more commonly than the adenohypophysis, and its destruction may result in diabetes insipidus. Hypopituitarism is uncommon, even when the adenohypophysis is involved, because of its large functional reserve.
B. Metabolic Diseases
- Langerhan’s Cell Histiocytosis. In this condition the neurohypophysis is frequently infiltrated with histiocytes and other inflammatory cells, interfering with neurosecretion. This is a significant cause of diabetes insipidus in children. Diabetes insipidus, punched out bone lesions and exophthalmos constitute the diagnostically useful triad for Hand-Schüller Christian Disease, a sub-type of Langerhan’s cell histiocytosis.
- Hurler’s Syndrome (Gargoylism). The adenohypophyseal cells contain large quantities of the mucopolysaccharides characteristic of this disorder.
- Other systemic disorders that may affect the pituitary include amyloidosis and hemochromatosis.
C. Inflammatory Processes
- Lymphocytic, xanthomatous and xanthogranulomatous hypophysitis are idiopathic chronic inflammatory conditions affecting adults, which may result in hypopituitarism. Up to one-third of affected patients, particularly those with lymphocytic hypophysitis, have autoimmune disorders affecting other endocrine and/or non- endocrine organs.
- The pituitary may also be affected by various systemic chronic inflammatory diseases, such as sarcoidosis.
D. Infections
The pituitary may be affected by contiguous infectious processes such as meningitis or sinusitis, or by septicemia. It may also be involved in syphilis, tuberculosis, and other systemic infections. Patients with AIDS may have a variety of pituitary infections.
Feedback/Errata